LA-SiGMA Projects
The description of each Science Driver (SD) project and the Cybertools and Cyberinfrastructure (CTCI)'s contribution is described below. For an overview of the SDs please visit the research section.
-
SD1, Focus 1:
Multiscale Methods for Strongly Correlated Materials
-
Investigators using the present set of numerical methods have begun to understand the individual phases of these systems. DFT, notably the Local Density Approximation (LDA), accurately describes a host of moderately correlated materials. A variety of augmentations ("LDA+" methods) allow DFT to address the role of stronger local correlations. A notable example is the combination of LDA or down folding LDA calculations with the Dynamical Mean Field Approximation (DMFA) and its cluster extensions, including the Dynamical Cluster Approximation (DCA). These methods have, for example, provided a qualitative understanding of the origins of antiferromagnetism and superconductivity in some systems. Yet these algorithms are fundamentally limited in their treatment of the different length scales. To understand the complexity that emerges due to the competition between different phases, a new suite of computational formalisms, algorithms, and codes is required. The SD team will attack the grand challenge problem of multiscale physics in strongly correlated systems by developing and applying novel methods that systematically incorporate nonlocal corrections to both LDA and DMFA.
The barriers that inhibit these developments are inherent to existing computational methods. DMFA and DCA map the lattice onto a cluster embedded in a self-consistently calculated effective medium. The cluster problem is usually solved using a Quantum Monte Carlo (QMC) simulation, while disorder can be included by averaging over configurations. Currently, either perfectly parallel (MPI) or hybrid parallel (MPI+OpenMP) calculations are used. For this project, in conjunction with the CTCI Novel Architectures team (Ramanujam), we will develop a hybrid continuous time QMC solver for 16-way multicore, hyperparallel computers such as NSF's Blue Waters. Nevertheless, calculations are limited by the "minus sign" problem, which is nonpolynomial hard. This means that simulations of correlated electrons take exponentially longer as temperature decreases and cluster size increases, making it very difficult to treat correlations on the important length scales. Another issue is that the LDA is an approximation based upon the numerical solution of the Homogeneous Electron Gas (HEG). Since the HEG does not have ordering or moment formation, it is difficult to describe these phenomena using LDA.
Significant developments beyond the state of the art are required to address these problems. For example, in the DMFA/DCA approach, another length scale must be introduced as in the Multiscale Many Body (MSMB) approach. This is accomplished by a multiple embedding scheme in which correlations over each length scale are treated with an appropriate approximation. Strong correlations at short length scales are treated with a numerically exact QMC simulation on a small cluster. This cluster is embedded in the larger cluster where the weaker correlations at intermediate length scales are treated using the parquet approximation. This larger cluster is embedded in an effective medium that is used to treat correlations on the longest length scales. For this project, we will develop an MSMB code capable of treating multiple correlated orbitals. For DFT, the Perdew group will develop a complementary approach to the strongly correlated electron problem that relies upon improved approximations for the exchange-correlation energy. A recent meta-GGA, which predicts accurate lattice constants and surface energies for solids and accurate atomization energies for molecules, is probably close to the limit of accuracy for semilocal functionals, and is a natural base on which to build the fully nonlocal approximations. A hyper-GGA currently under development based on these ideas, describes the strong correlation responsible for high energy barriers to chemical reactions, and also the moderate correlation present in ordinary matter. This functional will be incorporated into common DFT codes including VASP.
The payoff from these developments will be the greatly improved ability to study the correlated systems described in Foci 2 and 3, where overcoming the multiscale barriers will be transformative.
-
-
Focus 2:
Correlated Organic and Ferroelectric Materials
-
Studies of organometallic conductors and magnets will be performed using porphyrins and iron oxide clusters coated with biocompatible small-molecule capping ligands as testbeds. Porphyrins are excellent model systems for fundamental studies of the interrelationships between electronic properties and structure, due to their robust and versatile structural motifs, which allow production of a rich variety of molecular architectures. Molecular clusters formed by high-spin mid-3d-transition metals exhibit different forms of magnetic behavior, including ferromagnetism with slow relaxation rates.
Perdew, Ruzsinszky, and Burin (Tulane) and Derosa (LA Tech/Grambling) will study metalloporphyrin nanostructures to test and validate a wide range of DFT functionals, including the Local Spin Density Approximation (LSDA), the second generation GGA functionals, and the more recent meta- and hyper-GGA functionals developed by the Perdew group. Jarrell and Moreno (LSU) and Fishman (Oak Ridge National Labs) will use the calculated band structures to construct Hamiltonians that will be used in a DMFA/DCA study of the dynamic magnetic properties. These same Hamiltonian parameters will be used by Browne (LSU) and Derosa in nonequilibrium Green function calculations of charge transport in these systems. Garno (LSU) will prepare nanostructures of metalloporphyrins and perform magnetoresistance and electrical conductance measurements for comparison with and validation of the computational methods.
Burin, Ruzsinszky, and Perdew will use the new functionals to study high-spin molecules, such as iron oxide clusters (Fig. 1) embedded in organic molecules. To validate the computational approaches, Spinu (UNO) joined by Stevens and Goloverda (Xavier) will measure cluster magnetic properties using Squid Magnetometry techniques at UNO/ AMRI with samples synthesized by Kolesnichenko (Xavier).
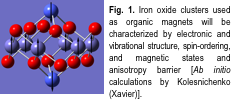
The complex emergent phenomena of multiferroic composites allow the interconversion of electric and magnetic energy. This is an emerging class of multifunctional materials for smart sensors, computer memories (utilizing both charge and spin to store and process information), and voltage-controlled magnetic devices such as tunable microwave filters. Malkinski and Caruntu (UNO/AMRI) and Meda (Xavier) will fabricate nanostructured multiferroic composites, and Whittenburg (UNO) will perform computer simulations using Perdew's GGA methods to predict the magnetic, ferroelectric, mechanical, magnetostrictive and electrostrictive properties and model devices constructed using these materials.
-
-
Focus 3:
Superconducting Materials
-
These methods will also be used to study the new pnictide and cuprate superconductors. The pairing mechanism in the pnictide materials has not been established. Proposals include phonons, correlation effects enhanced by nesting, or a more novel mechanism involving overscreening of the Fe-Fe interaction by As. Methods that combine LDA and MSMB/DCA will be used by Browne, Moreno, Vekhter, and Jarrell (LSU) and Bagayoko (SU) to study the first two mechanisms. Assessing the overscreening mechanism requires Perdew's new DFT methods, since conventional LDA will not capture the nonlocal effect of an As atom screening the interaction between adjacent Fe atoms, a major feature of the third mechanism. A central question surrounding the cuprates is: "What is under the superconducting dome?" surrounding a Quantum Critical Point (QCP) that was recently found in model calculations. The question of whether the order associated with the QCP, if any, competes with superconductivity must also be addressed. A new generation of massively parallel QMC and MSMB codes will be used to address these questions. For example, bottlenecks in the parquet equations include the contractions and rotations of large (greater than 100GB) rank three tensors (vertices), as well as numerical instabilities, which can be addressed with massively parallel linear systems solvers, such as BICG, BICGStab, or GMRES. These new codes will be developed by an extensive collaboration involving researchers at the Center for Computation & Technolgy CCT, including Ramanujam (the developer of advanced tensor rotation and contraction methods), Jarrell, and Moreno (LSU), together with Tomko (Ohio Supercomputer Center), Fishman (Oak Ridge National Laboratory), as well as the computational team. This team will focus on the calculation of thermodynamic and transport properties, which will be validated by Diebold and Mao (Tulane); and neutron and ARPES spectra, which will be validated by Plummer and DiTusa (LSU) and Sprunger and Kurtz (CAMD/LSU); all using samples made by Jin and Zhang at LSU.
-
-
SD2, Focus 1:
Electrochemical Capacitors and Fuel Cell Electrodes Based on Nanotube Forests
-
Fast, high-energy density, electrical energy storage materials will be a disruptive technological advance for effective utilization of intermittent and distributed power sources from the grid, for design of electrical vehicles, and for regenerative energy capture, including automobile braking. Pratt (Tulane) has carried out the first molecular simulations of proposed supercapacitors based on carbon nanotube (CNT) forests (Fig. 2). Zhao (SU) has promising preliminary results for the simulation-guided design of CNT-based fuel cell electrode materials. The goal of this focus area is to use novel MC and ab initio methods to overcome the multiple time scales barrier and study electrical storage materials. The payoff is the ability to design better electrochemical capacitors and fuel cells.

CNT forests are intrinsically multiscale problems. The interactions at the pore surfaces are 10-10 m scale and pore radii are ~10-9 m scale, while the height of the nanotube forest is 10-3 m. A research goal for capacitors is to improve the energy densities while retaining the natural advantages of capacitors of rapid response, long lifetimes, and temperature insensitivity. Molecular-scale simulations of these systems (Fig. 2) are feasible and yield single-electrode capacitances in the neighborhood of 80 F/g of electrode, in agreement with experimental capacitances of electrochemical capacitors utilizing carbon-nanotube forests or carbide-derived carbons as electrode material. The Alliance will extend this work to address a variety of practical issues.
Numerical simulations of pore filling will predict pore composition in important situations where reliable estimates are not available. This will characterize the dynamics over the relevant range of 10-9 s to 10-3 s for candidate materials. Ashbaugh, Pratt, and Bishop (Tulane) will develop and apply (with CTCI help) MC algorithms applicable to phase equilibria. Current models assume that a quantum capacitance adds in series with the electrochemical capacitance. The Alliance will use DFT methods to test this assumption using atom-level charging of graphite electrodes. Coarse-grained electron density models will be implemented (with CTCI guidance) in large-scale MD simulations. Pesika (Tulane) will carry out cyclic voltammetry on nanotube forests to explore the potential dependence of the experimental capacitances. The stored energy depends quadratically on the electric potential, and operation at potentials as high as 4V could achieve a high energy density. But extraneous chemical damage must be avoided. Incorporation of chemical processes in molecular simulation is at the forefront of materials simulation research. Rick (UNO) will incorporate the reactive impurities from H2O redox chemistry to simulate damage expected at high potentials.
Zhao and Bagayoko (SU) will use DFT-MD with Perdew functionals to understand the catalytic mechanism of the oxygen reduction reaction at nitrogen-doped CNTs. Preliminary simulations show that N edge-doped CNTs are the most stable structures. Henry (SU) will build fuel cells with these materials and will evaluate the effectiveness of the new designs. The CTCI group will port these calculations to next generation computers.
-
-
Focus 2:
Thermodynamics and Kinetics in H2 Storage Systems
-
Among the barriers that hinder the use of hydrogen as a clean alternative to hydrocarbon fuels are absorption/desorption rates, volume/weight ratios, hydride stability, and desorption temperatures of current H2 storage materials. The Alliance will use novel multiscale MD and MC simulations, kinetic MC, finite element, and finite difference modeling to predict rates of hydrogen uptake/release over time scales reaching 103 s and extending over several microns for metal alloy storage materials with new catalytic additives. Other Alliance members use X-ray tomography to probe these materials over the same length and distance scales. The goals of this focus area are to predict the influence of catalytic additives in enhancing atomic mobilities and desorption rates in metal hydrides and to explore a wider range of potential hydrogen storage materials. The payoff will be an improved ability to design materials for hydrogen storage.
Ab initio molecular scale energetic, structural, dynamic properties will be calculated by Mainardi (LA Tech). Wick (LA Tech), Chen and Hall (LSU) will develop force fields appropriate for these reactive systems to supplement the ReaxFF force fields. Hall, Chen, Wick, and Mobley (UNO) will use advance ensemble methods to calculate the free energies of different compositions and to determine the spinodal points of mixtures. Mainardi and Dai (LA Tech) will study diffusion and reactions using kinetic MC and employ finite element and finite difference methods for extended length and time scales. CTCI group members will port codes, allow data reuse, and provide efficient work flow. Butler (LSU) and Dobbins (LA Tech/Grambling) will employ the LSU X-ray synchrotron tomography beamline and the Argonne Advanced Photon Source nanotomography beamline to study in situ and ex situ de/rehydrogenation reactions, to assess 3D microstructures and interphase boundaries, and to quantify the distribution and size of single phase domains. The CTCI visualization toolkits will be utilized for this part of the project. Since atomic diffusion rates may limit the desorption, Browne (LSU) will use Fick's Law diffusion and non-equilibrium chemical models to model solid state diffusion and phase transitions. The temperature-dependent diffusion coefficients will be determined using diffusion and diffusion-reactions equations and experiments. The results of these simulations, calculations, and experiments will be the prediction of optimum conditions and materials for hydrogen release and uptake.
-
-
Focus 3:
Catalytic Reactions Involving Metal Oxides
-
Metal oxides are an important class of catalytic materials used in industry and are implicated in the formation of hazardous materials when formed in the environment. The scarcity of accurate force fields is the barrier that limits modern simulation for these materials. The goal of this focus area is to develop novel, reliable and transferable reactive force fields incorporating polarizabilities and environment dependent atomic charges. The potential parameters will be determined from structural calculations on large systems and charges, and their fluctuation parameters will be calculated from fits to electrostatic potentials. Massively parallel implementations (guided by the CTCI teams) of modern DFT functionals will be used to incorporate the force fields into the calculations of the SD teams. The payoff will be the ability to design better metal oxide catalysts and force fields for use in the other SDs.
Burning lignocellulosic biomass (e.g., wood chips) in low oxygen conditions yields water gas, a mixture of CO and H2O. The water gas shift reaction is used to obtain CO2 and H2 from this mixture. The H2 can then be used as a source of energy. Experimentalists at LA Tech and Grambling have perfected methods for synthesizing metal oxide catalysts that promote this reaction under various conditions. Experimentalists at LA Tech have developed novel nanostructured catalysts that convert water gas to higher hydrocarbons or alcohols using Fischer-Tropsch processes, while avoiding some of the well-known problems with tar formation. Despite their importance, a molecular-level understanding of the catalytic processes in these two reactions is lacking. Complicating their utility, when metal oxide nanoparticles are released into the environment (through, for example, incineration of spent catalysts), they can catalyze production of Polychlorinated Dibenzo-p-Dioxins (PCDDs) and Dibenzofurans (PCDFs).This focus area will study both the beneficial and harmful features of metal oxides.
Rick (UNO), Ramachandran and Wick (LA Tech), and Hall and Chen (LSU) will lead the computational effort using DFT, force field methods, and MC/MD simulations. Siriwardane (LA Tech) and Seetala (Grambling) will synthesize catalysts and use scanning probe microscopy and X-ray diffraction methods to study catalyst structure. They will evaluate catalyst performance using flow and batch reactors coupled to gas chromatographs. The computational and experimental team members will work closely, with shared students, to validate the computational models and force fields for metal oxides. Since catalytic processes nearly always involve bond breaking, atomic/molecular migration, and bond formation at different active sites at different times, the ability to deal with multiple length and time scales is essential. The validated models will be used to explore the possibilities of improving catalyst performance, and promising candidates will be examined by Siriwardane and Seetala. These studies will directly benefit two start-up companies in Ruston (Renewable Fuels, LLC, and Carbon Capture Energy Technologies, Inc.) that have provided letters of commitment for this work.
The initial steps in the gas phase formation of PCDDs and PCDFs, and the structure and reactivity of small copper oxide clusters are now being studied to understand this molecular process. Accurate metal oxide force fields and sophisticated MC methods are needed to simulate the larger cluster sizes that are being studied experimentally by Dellinger (LSU). Hall and Chen will use the metal oxide force fields to study the structures and reactivities of metal oxide clusters containing 20-30 metal atoms, and to compare to the experimental work of Dellinger. Zhao and Bagayoko (SU) will test the predicted structures and reactivities using DFT-MD.
-
-
SD3, Focus 1:
Unimolecular vehicles
-
Modern polymerization techniques can precisely synthesize nanoscale polymer components. Efficient coupling reactions, such as the Huisgen "click" reaction, permit individual polymeric units to be linked to larger, modular assemblies that can be built into supramolecular structures for drug and peptide encapsulation that improve their solubility and/or stability in vivo. Grayson (Tulane) will synthesize and characterize a modular library of core molecules (Fig. 3) and amphiphilic side chains (including pH sensitive and biodegradable functionalities) to explore encapsulation based on architecture and chemistry. Architectures to be synthesized include linear, star, dendrimer, and macrocycle topologies. Encapsulation and hydrophobic dye (pyrene) solubilization in water will be tested using UV-vis. Light scattering will verify the size of the host-guest complexes. Dye-labeled hydrophobic peptide encapsulation will also be investigated. The goal of this focus area is to design better unimolecular encapsulation materials.
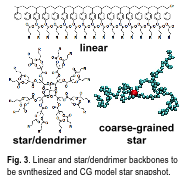
Ashbaugh and Bishop (Tulane) will explore the role of architecture and solvent to optimize vehicles for controlled capture and release using hybrid, CG, and acceleration strategies. Molecular simulations will analyze drug solubility in aqueous/nonaqueous solvents and polymer side chain-guest interactions. Poly(ethylene oxide) oligomers will serve as a prototypical hydrophilic side chain, while poly(caprolactone) and polystyrene oligomers will serve as prototypical hydrophobic side chains. Guests to be studied include pyrene and drug peptides. Novel inverse MC simulations will be used to develop CG models (to overcome the time scale barrier) whose intra/intermolecular correlations match those of the molecularly detailed simulations described previously. Simulations will allow assessment of the effect of molecular topology (Fig. 3) on encapsulation efficacy. Depending on the topology, different free energies for absorbing/delivering cargo in differing environments will be used to optimize transport efficacy. Computed solubilities will be benchmarked against experiment, and molecular topologies studied by simulation will narrow synthetic targets. A second unimolecular delivery vehicle, halloysite nanotubes, will be studied computationally by Derosa (LATech/Grambling) and experimentally by Lvov (LA Tech). Preliminary experimental work has established these nanotubes as effective sustained delivery vehicles. The payoff: experimentally validated, multiscale models will be developed to predict drug transport from halloysite nanotubes.
-
-
Focus 2:
Self-assembled delivery vehicles
-
As a complement to unimolecular carriers, drugs can be entrapped in surfactant assemblies with dimensions less than 100 nm and absorbed via paracellular and transcellular routes in the intestine at rates dependent on the nanoparticle size, surface charge, and hydrophobicity. The goal of this focus area is to combine novel MD and CG simulation and experimental studies to examine the effects of nanoparticle properties on translocation efficacy through cell membranes. To overcome the time scale barrier, both all-atom and CG large-scale MD simulations will be conducted by Moldovan and Nikitopoulos (LSU) and Bishop (Tulane) using GROMACS and LAMMPS to investigate the mechanism(s) of early stage surfactant self-assembly and the release/translocation of drugs through lipid bilayers (Fig. 4). The MD and CG investigations by Moldovan, Nikitopoulos, Bishop, and Ashbaugh are critical for understanding the mechanisms of drug absorption. They can serve to design experiments and to discriminate between mechanisms. The payoff: the determination of the optimal conditions (i.e., concentration, pH) under which desired structures are stabilized will be used experimentally to build delivery systems with functionalities targeted to specific applications.

Experimental validation involves the synthesis and characterization of nanoscale delivery systems for improved bioavailability via interfacial phenomena such as the formation of micelles and vesicles, and transport of nanostructures across lipid bilayer membranes, thus emulating cellular uptake. Surfactants and polymers will control nanostructure and functionality. Sabliov (LSU) will use emulsion evaporation to synthesize chitosan/poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles with entrapped α-tocopherol (Chi/PLGA(αT)). The nanoparticles morphology, size distribution, and zeta potentials will be characterized using dynamic light scattering and TEM. The passive transport of Chi/PLGA(αT) nanoparticles through phospholipid bilayers will be investigated using both MD and CG simulations. A biomimetic artificial membrane permeation assay based on dipalmitoyl-phosphatidylcholine liposomes will be used to experimentally assess passive in vitro transport of Chi/PLGA(αT). The mechanistic insights revealed by the MD and CG studies will impact directly our fundamental understanding of the role of the Chi/PLGA(αT) nanoparticle characteristics on their transmembrane permeability, ultimately leading to improved delivery protocols.
-
Collapse All
Expand All